Orogen: Unweighted by Population | DNA Science
The only relationship predictions based on data from a peer-reviewed source
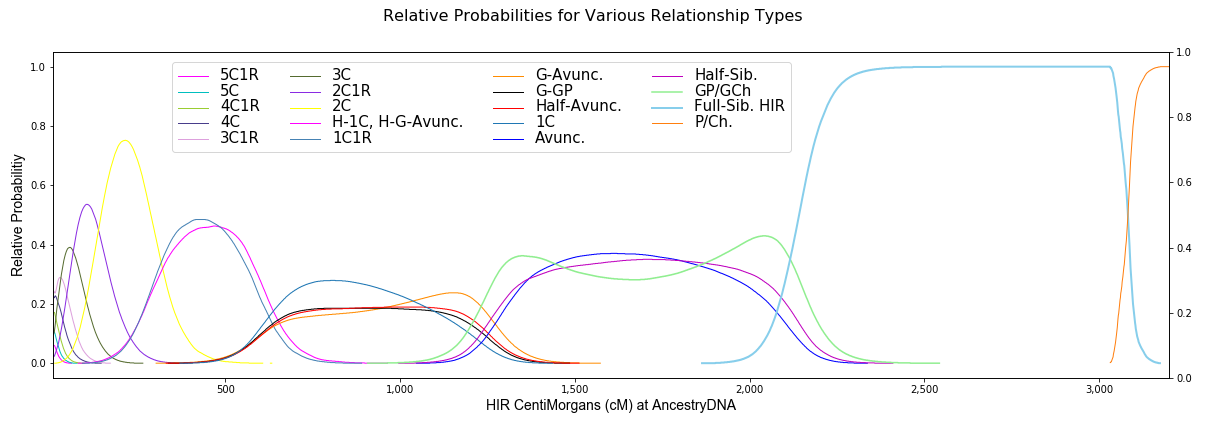
This tool calculates probabilities using the most accurate shared DNA data available (from Ped-sim). The relationship prediction tools at this site are the first to take into account differences between maternal and paternal relationships and show that close genealogical relationships considered to be in the same group are sometimes quite different.
Enter your shared cM in the applicable box below
For a multiple cousin relationship predictor, click here. For relationship probabilities weighted by proportion in the population, click here. You would want to use the weighted predictor if you have no idea who your match is. You would want to stay on the current page if you believe you know how your match fits into your family tree and you're checking to confirm that. Population weights give a significant advantage to more distant cousins because a person likely has about 5x as many cousins from one generation of ancestors compared to those from the next generation of more recent ancestors.
1C1R = 1st cousin, once removed; cM = centiMorgan; HIR = half-identical regions; IBD = identical by descent (HIR + FIR).
All probabilities are for autosomal DNA only. Please subtract any X-DNA before using the calculator.
The above probabilities assume no endogamy or other pedigree collapse. Those cases should be treated separately.
*Parent/child and full-sibling relationships are easy to distinguish from each other or any other relationships. Parent/child relationships consist of a half-identical match across the whole length of the genome. Full-siblings share 12.5% fully-identical regions (FIR), on average. Genotyping sites will take this into account in their relationship predictions. Trust the labels given at the original testing site if both people have their DNA there.
Probabilities are included for relationships as far back as 8C1R. The huge advantage of this tool, other than the accuracy of the data, is that it treats close relatives as not being in the same group because the curves are significantly different. For distant relatives, there's much less certainty about the genealogical relationship for your DNA matches. Matches as low as 8 cM are allowed here, however the relationship may be farther back than 8C1R. While the relative probabilities are accurate for the relationship types shown, one also has to consider that the relationship is farther back. Indeed, any of the probabilities shown above are only relative to the other relationships listed, therefore they’re only meaningful in comparison to the other relationships. And there's no cM value at 8 cM or above at which even a 4C1R is the most probable relationship. So, while the probability of an 8 cM match may be higher for "4C1R or more distant," listing each relationship type separately would not result in more useful information. Not only are very low cM values difficult to assign to a recent ancestor, but segments of 20 cM or 30 cM may be on pile-up regions and therefore come from very distant ancestors.
Totals will not always add up to 100%. When more relationship types are possible, the chances of rounding errors increases. For more information about the methodology and discoveries associated with this tool, click here.
The data used for these predictions came from Ped-sim. In this case, the refined genetic map of Bhérer et al. (2017) was used as well as the crossover interference parameters of Campbell et al. (2015).
Note that the probability curves in the plot below are very similar to those published here in April, 2021.